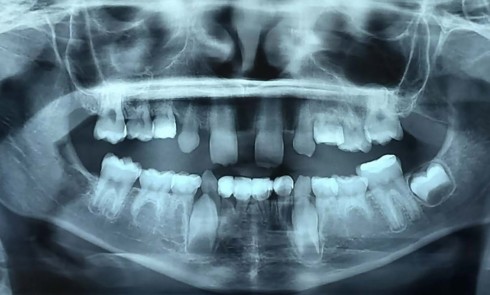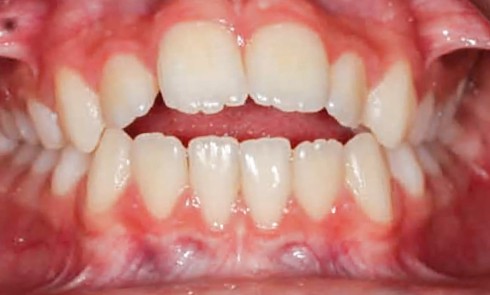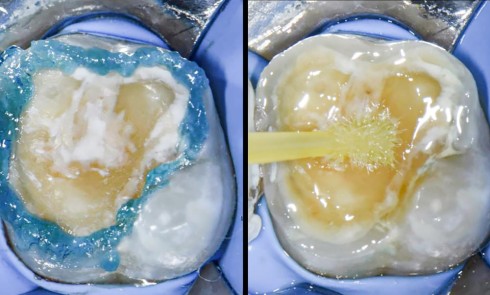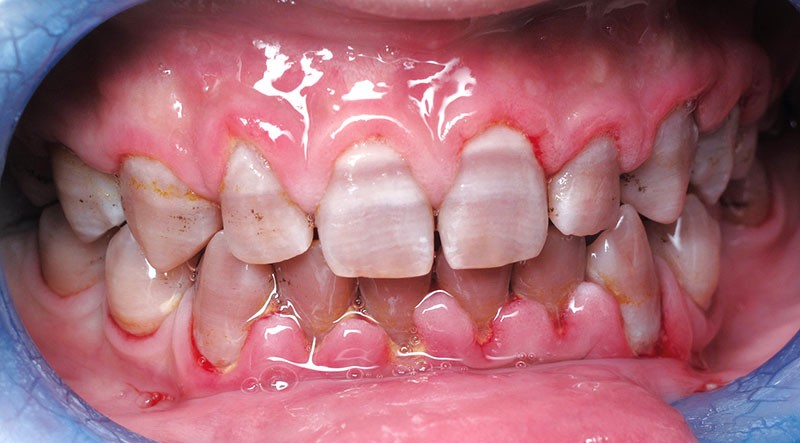

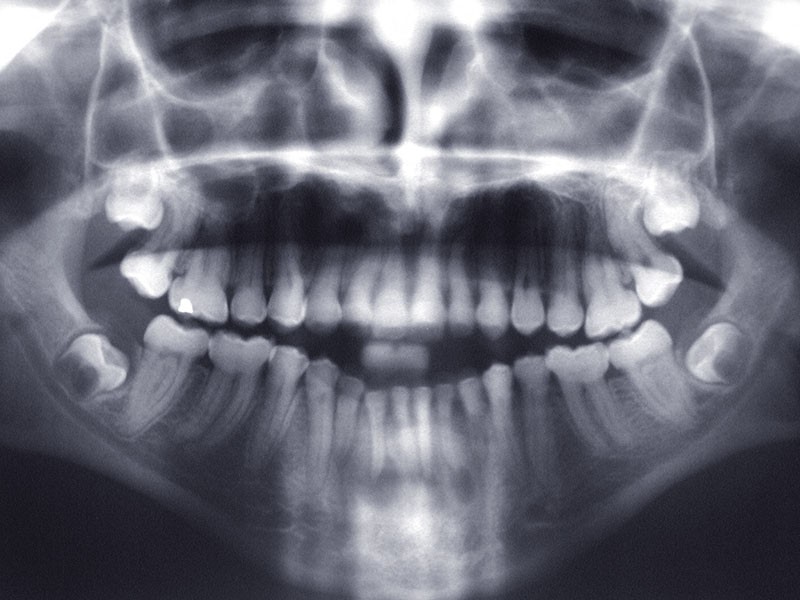
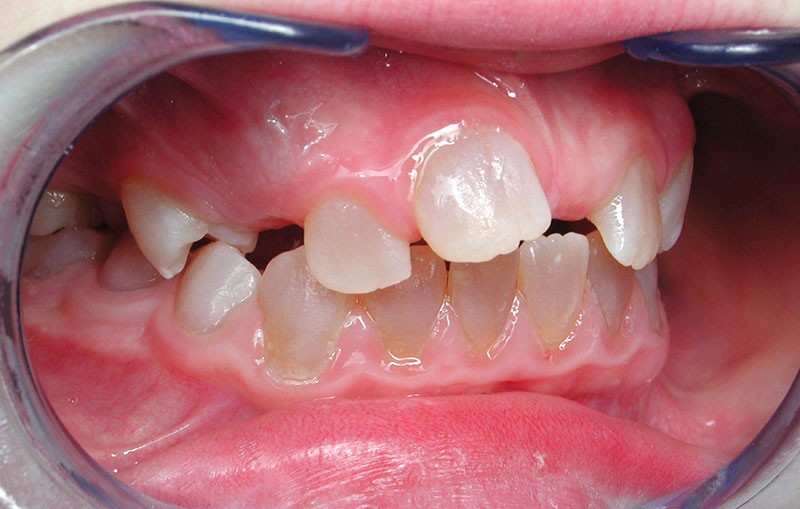
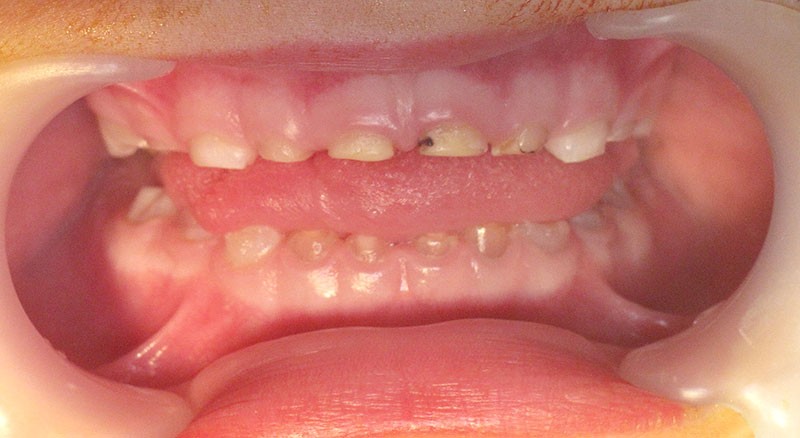
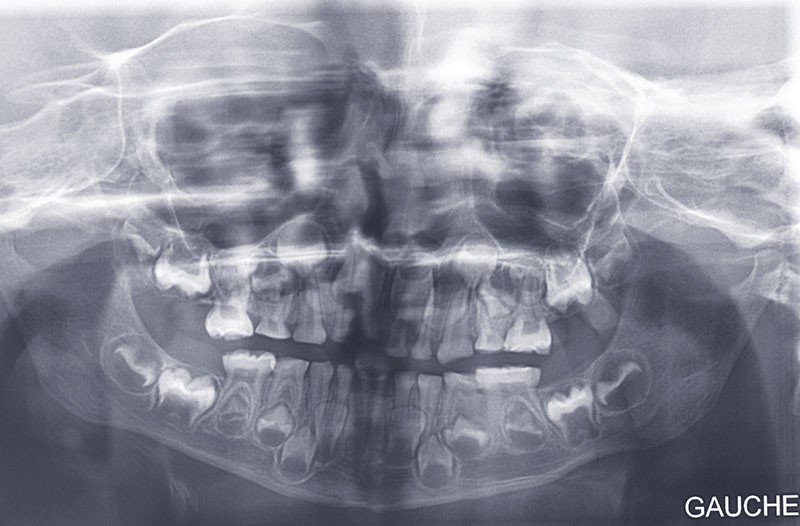
Teinte opalescente grise à jaune ambrée, couronnes globuleuses, calcifications intra-pulpaires, racines fines et courtes, attrition précoce, nécrose aseptique… Ces anomalies cliniques et radiographiques décrivent le tableau de la dentinogenèse imparfaite (DI) isolée de type 2. Cette anomalie génétique à transmission autosomique dominante est liée à un défaut de DSPP, gène de la sialophosphoprotéine dentinaire [1, 2]. Des anomalies dentinaires similaires résultent également de mutations de ce gène. Les avancées en recherche génétique tendent à réunir ces pathologies sous une même étiologie.
Un diagnostic précoce est primordial, car il existe aussi une forme associée à une anomalie osseuse, l’ostéogenèse imparfaite. Le chirurgien-dentiste est donc au premier plan pour dépister ces anomalies et limiter dès le plus jeune âge leurs conséquences infectieuses, fonctionnelles et esthétiques.
Diagnostic clinique
Shield [3] a proposé en 1973 une classification fondée sur des différences phénotypiques. Il distingue les anomalies dentinaires isolées des anomalies dentinaires syndromiques (tableaux 1 et 2).


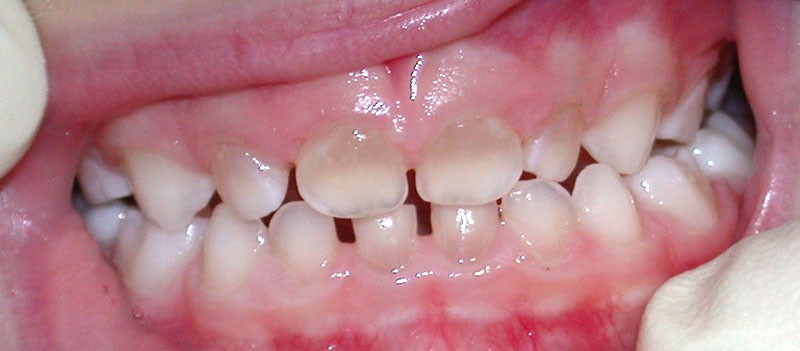
Diagnostic génétique
Cliniquement, certains cas peuvent présenter des caractéristiques appartenant à plusieurs sous-groupes de la classification de Shield, rendant le diagnostic précis difficile [20, 21]. Cette classification est donc obsolète depuis les progrès des analyses moléculaires du génome.
L’analyse génétique montre que les phénotypes de ces anomalies dentinaires isolées résultent de mutations du gène de la DSPP. Ce gène, porté par le bras long du chromosome 4, code pour une protéine secrétée par les odontoblastes et les pré-améloblastes. Elle est également présente dans le cément et l’os…