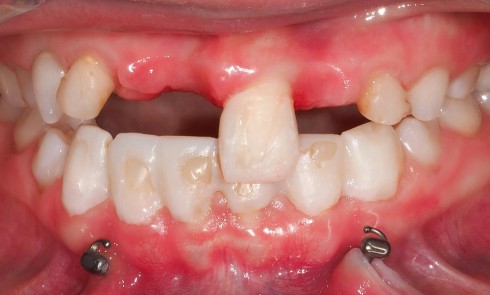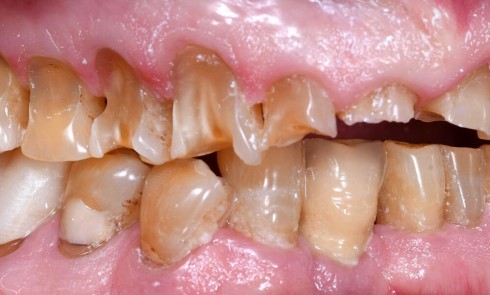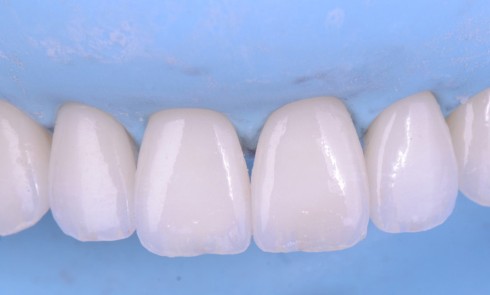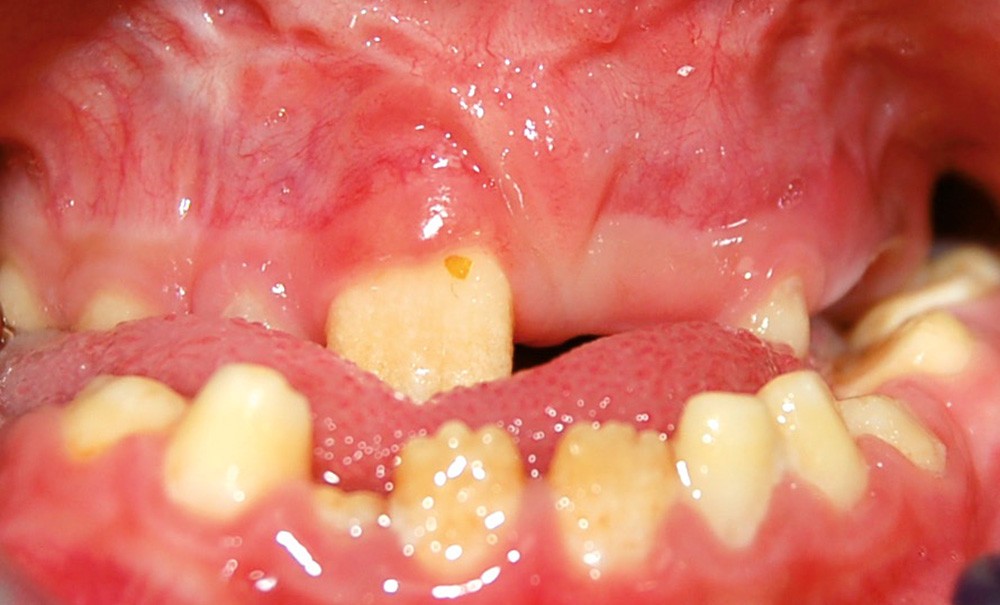

Les anomalies de structure des tissus minéralisés amélaires constituent un groupe d’affections bucco-dentaires hétérogène sur les plans phénotypiques et moléculaires. Ces anomalies du développement peuvent être isolées, comme les amélogenèses imparfaites (AI) avec mutations dans les gènes AMELX ou ENAM, ou syndromiques, dans le cadre d’associations plus complexes avec des atteintes systémiques pouvant impliquer, entre autres, les systèmes neuro-sensoriels, squelettiques, génito-urinaires ou cardio-vasculaires.
L’objet de cette publication est de décrire les principales formes d’AI syndromiques, leurs aspects cliniques et génétiques, ainsi que les stratégies de prise en charge.
Les voies de signalisation impliquées dans les cytodifférentiations améloblastiques, la sécrétion, la maturation, ainsi que la biominéralisation matricielles sont pléiotropiques et jouent un rôle essentiel durant le développement, ce qui explique les associations entre anomalies dentaires et autres pathologies générales. Les processus de transport protéiques et ioniques (gènes WDR72, SLC13A5 ou SLC24A4 par exemple), d’adhésion cellule-cellule et cellule-matrice (gènes ITGB6, LAMA3 ou LAMB3 par exemple), de dégradation protéasique (MMP20 ou KLK4 par exemple) ou de pH sensing (GPR68, par exemple) peuvent également être altérés dans certaines formes isolées et syndromiques d’anomalies de structure [1].
Ces contextes syndromiques nécessitent un bilan exhaustif et une prise en charge médico-chirurgicale multidisciplinaire. Les centres de référence et de compétence maladies rares coordonnent l’équipe de spécialistes et centralisent les informations relatives aux aspects diagnostiques et thérapeutiques.
Formes d’amélogenèse imparfaites (AI) syndromiques
Le syndrome émail-rein
Le syndrome émail-rein ou ERS (enamel renal syndrome) (OMIM 204690, ORPHA 1031), également appelé AI avec néphrocalcinose, est caractérisé par…